TAFFISH flow page
TAFFISH 流程主页
phylogeny-flow
A compact, inspectable phylogenetic analysis route for homologous protein or DNA sequences: multiple
sequence alignment, optional trimming, tree inference, static tree visualization, report, and provenance.
面向同源蛋白或 DNA 序列的紧凑、可检查系统发育分析路线:多序列比对、可选修剪、建树、静态树图、报告和溯源记录。
Example data
示例数据
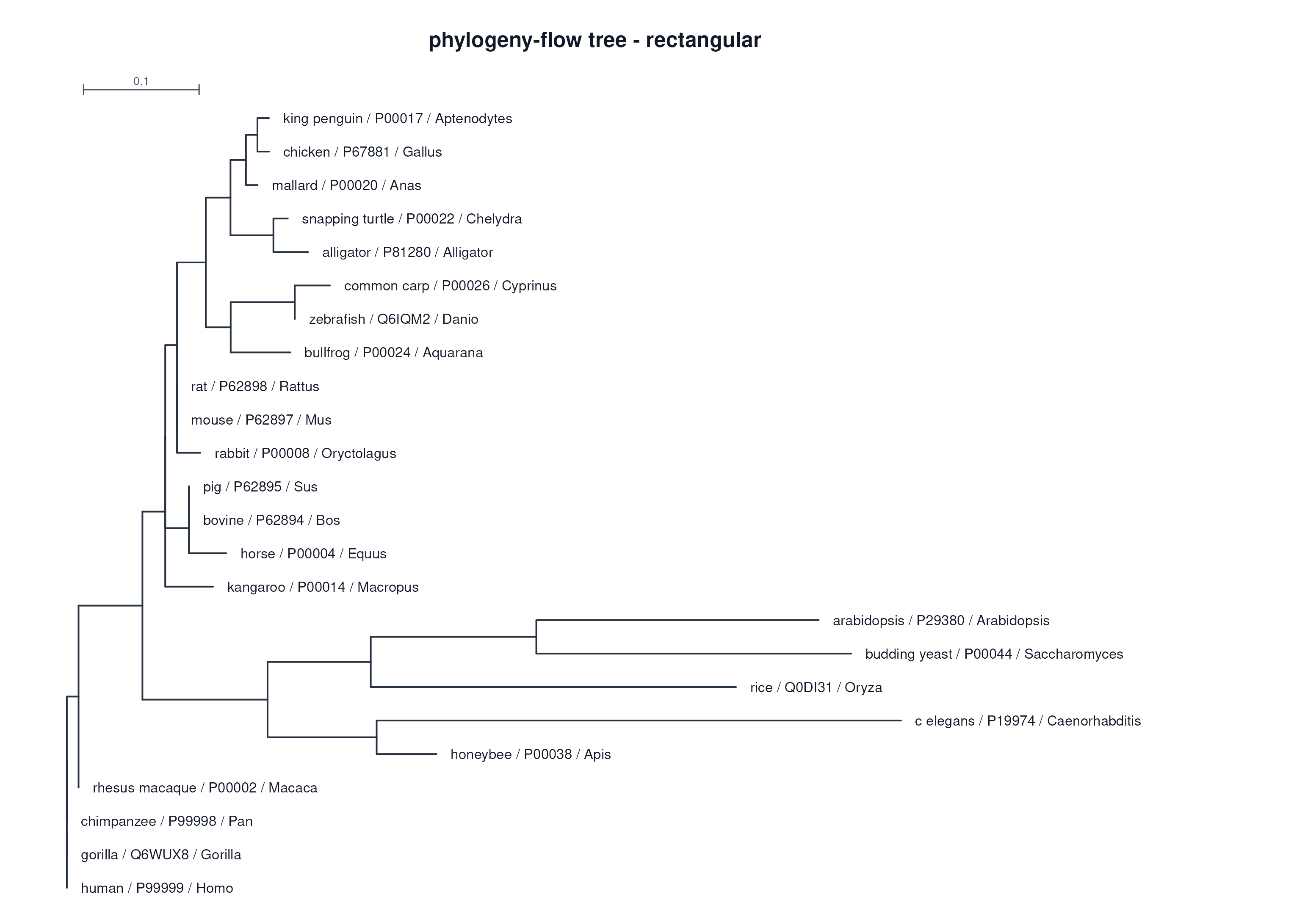
24 cytochrome c proteins
24 条 cytochrome c 蛋白序列
Default route
默认路线
MAFFT -> trimAl -> IQ-TREE
Tree model
建树模型
LG
Report status
报告状态
bilingual static HTML
双语静态 HTML
Route
流程路线
The flow is intentionally narrow: it starts from sequences that are already homologous and keeps the
command record transparent.
这个流程有意保持边界清楚:它从已经确认同源的序列开始,并保留透明的命令记录。
Input FASTA
输入 FASTA
Homologous sequences
同源序列
MSA
多序列比对
MAFFT / MUSCLE / Clustal Omega
Trimming
比对修剪
trimAl / ClipKIT / none
Tree
建树
IQ-TREE / FastTree
Report
报告
Plots, methods, versions
树图、方法、版本
Inputs
输入
Use raw homologous FASTA with --input, or provide a pre-aligned FASTA with
--alignment.
可以用 --input 传入同源 FASTA,也可以用 --alignment 传入已比对 FASTA。
Outputs
输出
Alignment FASTA, trimmed alignment, Newick tree, rectangular/circular tree plots, HTML report, commands,
versions, methods, logs, and manifest.
输出比对 FASTA、修剪后比对、Newick 树、矩形/环形树图、HTML 报告、命令、版本、方法记录、日志和运行记录。
Scientific boundary
科学边界
The final tree is a gene/protein tree for the chosen sequence set. It does not choose homologs,
determine orthology, or automatically become a species tree.
最终树是给定序列集下的基因/蛋白树。它不负责选择同源序列、判定直系同源,也不自动等同于物种树。
Parameter Guide
参数说明
Most runs only need an input, an output directory, and a small number of route choices. The options below
are the stable user-facing controls exposed by the flow.
大多数运行只需要输入、输出目录和少量路线选择。下面列出的是这个流程面向用户稳定暴露的主要参数。
--input PATH
--alignment PATH
Use --input for unaligned homologous FASTA; use --alignment when you already trust an aligned FASTA.
--input 用于未比对的同源 FASTA;已有可信比对时用 --alignment。
They are mutually exclusive. Existing-alignment mode records aligner=none.
二者不能同时使用;已比对输入会记录 aligner=none。
--outdir PATH
--force
Choose a dedicated output directory for one run.
为一次运行选择专门的输出目录。
Existing directories are refused unless --force is set.
默认拒绝已有目录;确认覆盖时才使用 --force。
--seq-type auto|dna|protein
Tell the flow whether the sequences are DNA or protein, or keep automatic detection.
指定序列是 DNA 还是蛋白;也可以保留自动判断。
Default is auto. Set it explicitly when the input is short or ambiguous.
默认是 auto;输入较短或容易误判时建议显式指定。
--aligner mafft|muscle|clustalo|none
Select the multiple-sequence aligner for raw FASTA input.
为未比对 FASTA 选择多序列比对工具。
Default is mafft. Existing-alignment input forces none.
默认是 mafft;已比对输入会强制使用 none。
--trimmer trimal|clipkit|none
Choose whether and how to trim the alignment before tree inference.
选择建树前是否修剪比对,以及使用哪种修剪工具。
Default is trimal. Use none when preserving every column matters.
默认是 trimal;需要保留全部列时使用 none。
--tree-engine iqtree|fasttree
Choose the tree inference engine.
选择系统发育树推断工具。
iqtree is the stricter default; fasttree is faster and approximate.
iqtree 是更严格的默认路线;fasttree 更快但更近似。
--model MODEL
Set the IQ-TREE model, or use automatic model selection.
设置 IQ-TREE 模型,或使用自动模型选择。
Default is auto. The cytochrome c example uses LG.
默认是 auto;cytochrome c 示例使用 LG。
--bootstrap N
--alrt N
--seed N
Control IQ-TREE support values and reproducible random choices.
控制 IQ-TREE 支持率计算和可复现随机选择。
Defaults are 1000 for UFBoot and SH-aLRT; use 0 to disable in fast demos.
UFBoot 和 SH-aLRT 默认都是 1000;快速演示时可设为 0。
--plot-tree true|false
--plot-layout rectangular|circular|fan
--plot-formats pdf,png,svg
Control static tree visualization and the report gallery.
控制静态树图和报告图集。
The default report keeps rectangular and circular views; fan is kept when selected as the primary layout.
默认报告保留矩形和环形视图;扇形图只在设为主布局时保留。
--sanitize-ids true|false
--threads N
Normalize sequence IDs and set the thread count for supported tools.
规范化序列 ID,并设置支持多线程工具的线程数。
ID normalization writes 00_inputs/sequence_id_map.tsv; default thread count is 4.
ID 规范化会写出 00_inputs/sequence_id_map.tsv;默认线程数为 4。
Real Example Report
真实示例报告
This embedded report is copied from the latest local example output and published under the
cytc-out path. It is the same static artifact a user opens after running the flow.
下面嵌入的报告来自最新本地示例输出,并发布在 cytc-out 路径下,也就是用户运行流程后会打开的静态结果。
Run Locally
本地运行
Install from the Hub index, inspect help, then run with an explicit output directory.
从 Hub 索引安装,先看 help,再用显式输出目录运行。
taf update
taf install phylogeny-flow
taf-phylogeny-flow --help
taf-phylogeny-flow \
--input sequences.fa \
--outdir phylogeny-out \
--model LG \
--threads 4
Good flow pages should show both the stable interface and a real report artifact. GitHub remains the source
code home, but the public website should be the first reading surface for users who want to understand what a
TAFFISH flow does.
好的流程主页应该同时展示稳定接口和真实报告产物。GitHub 仍然是源码入口,但官网应该成为用户理解一个
TAFFISH 流程做什么、产出什么、如何阅读结果的第一阅读面。
{kind=link}