Input data and parameters
Input
| Analysis date: | Thu May 28 10:47:47 CST 2026 |
| BAM file: | TAFFISH_RNASEQ_EXAMPLE_ROOT/yeast-standard-out/03_results/alignment/03_results/bam/SNF2KO_06.sorted.bam |
| Counting algorithm: | uniquely-mapped-reads |
| GTF file: | TAFFISH_RNASEQ_EXAMPLE_ROOT/yeast-standard-out/03_results/alignment_qc/00_inputs/genes.gtf |
| Number of bases for 5'-3' bias computation: | 100 |
| Number of transcripts for 5'-3' bias computation: | 1,000 |
| Paired-end sequencing: | no |
| Protocol: | non-strand-specific |
| Sorting performed: | no |
Summary
Reads alignment
| Number of mapped reads: | 482,516 |
| Total number of alignments: | 542,226 |
| Number of secondary alignments: | 59,710 |
| Number of non-unique alignments: | 103,019 |
| Aligned to genes: | 361,313 |
| Ambiguous alignments: | 72,171 |
| No feature assigned: | 5,723 |
| Not aligned: | 17,484 |
| Strand specificity estimation (fwd/rev): | 0.51 / 0.49 |
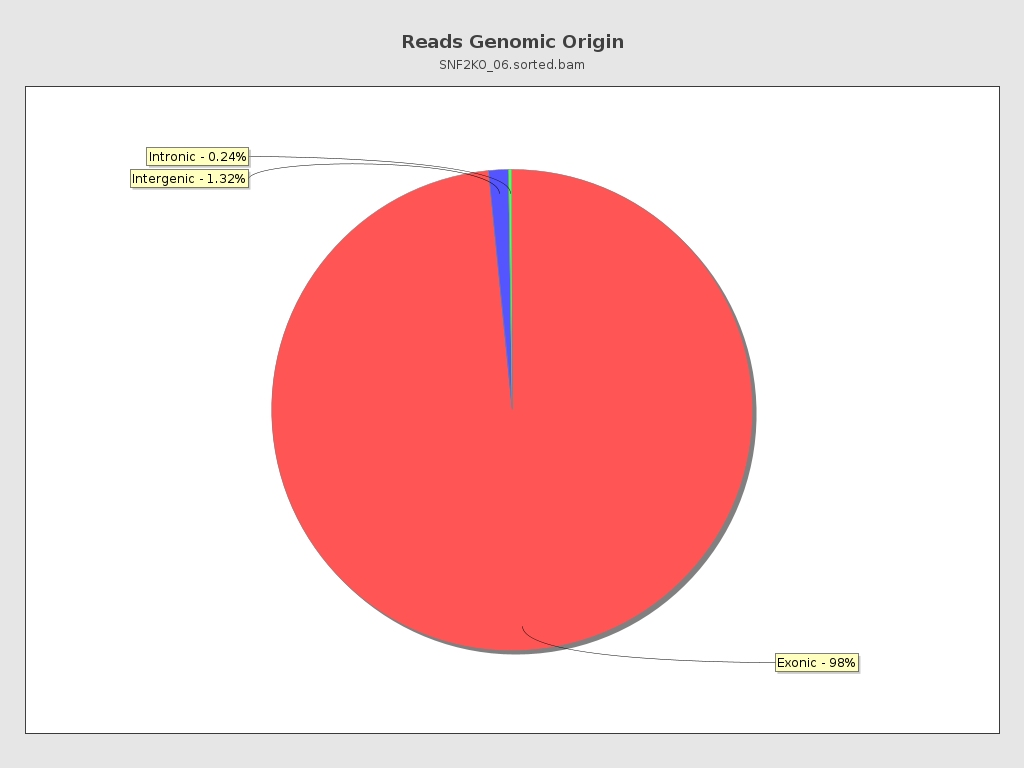
Reads genomic origin
| Exonic: | 361,313 / 98.44% |
| Intronic: | 859 / 0.23% |
| Intergenic: | 4,864 / 1.33% |
| Intronic/intergenic overlapping exon: | 15,151 / 4.13% |
Transcript coverage profile
| 5' bias: | 0.34 |
| 3' bias: | 0.2 |
| 5'-3' bias: | 1.7 |
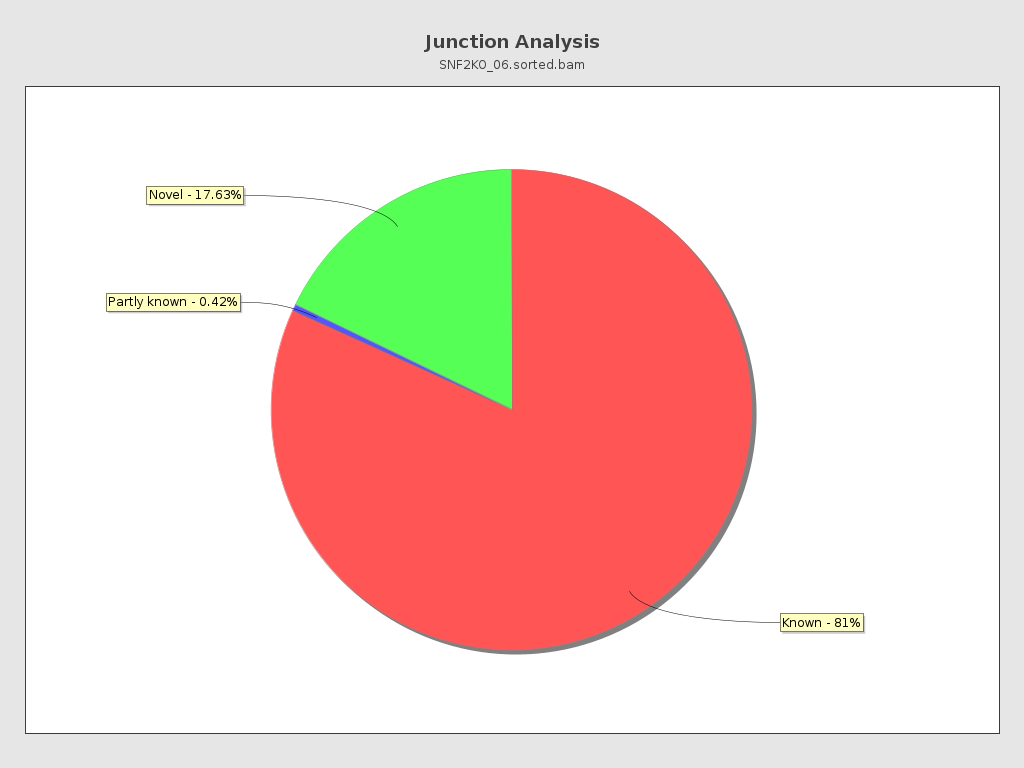
Junction analysis
| Reads at junctions: | 4,952 |
| TGCT | 4.14% |
| CTCT | 3.39% |
| AAAT | 2.97% |
| TGGC | 2.95% |
| ACCA | 2.89% |
| AGGT | 2.75% |
| CTGC | 2.67% |
| TCAC | 2.61% |
| AGAC | 2.44% |
| AACC | 2.38% |
| TAGT | 2.34% |
.png)
.png)
.png)